Adenosine deaminase deficiency

Results in accumulated ATP and prevents DNA synthesis. Causes SCID
Lesch-Nyhan syndrome

Absent enzyme: HGPRT in the purine salvage pathway. You get excess uric acid production. Self-mutilation, retardation, aggression, gout.http://www.youtube.com/watch?v=bxrCYhCyCYs
Osteogenesis imperfecta
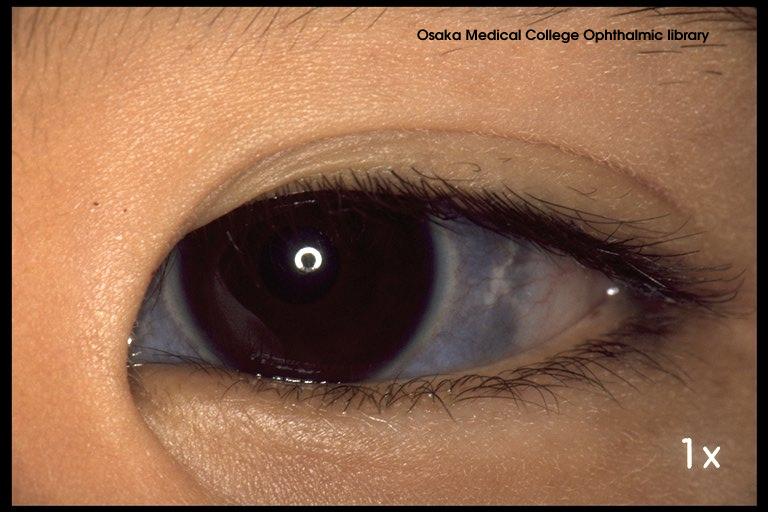
Brittle bones due to genetic defect. Most common if autosomal dominant and effects Type 1 collagen. Patient will have blue sclera (whites of the eyes are blue), hearing loss, and teeth will look bad.
Prader-Willi syndrome
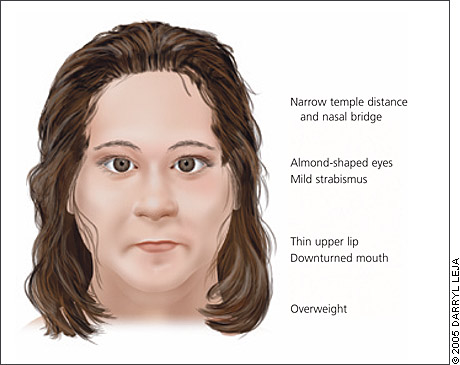
Paternal chromosome 15 is not passed down.
Angelmans syndrome

Loss of maternal contribution of chromosome 15.
Achondroplasia
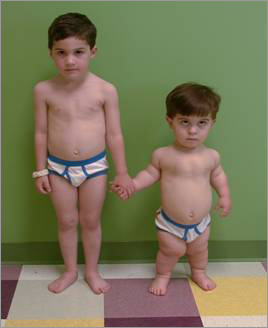
Defect in FGF receptor 3. Associated with advanced paternal age. Dwarfism with short limbs and larger head. Auto-dominant.
Huntingtons disease
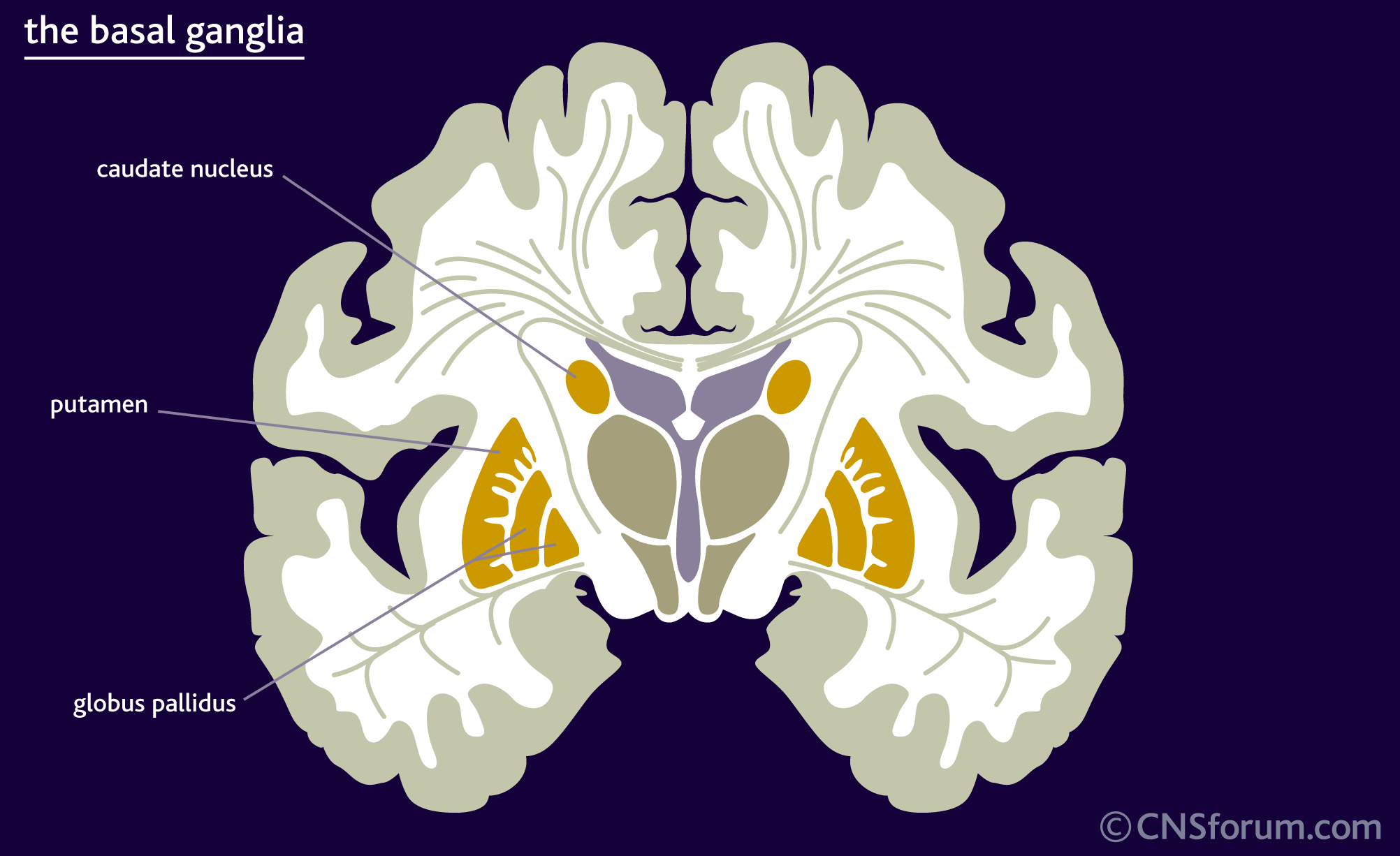
Disorder found on chromosome 4. Manifests between the ages of 20 and 50. Causes dementia, choreiform movement, caudate atrophy. Low levels of GABA and ACh in brain. Auto-dominant.
http://www.youtube.com/watch?v=QjPnzNUAm7E
http://www.youtube.com/watch?v=QjPnzNUAm7E
Marfans syndrome
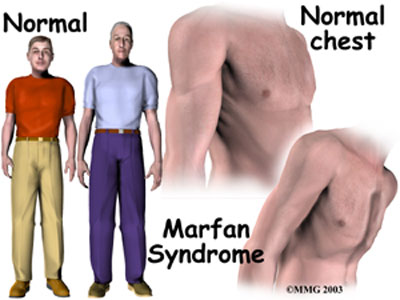
Connective tissue disorder. Some cardiac problems may be found such as a floppy mitral valve, aortic incompetence, and dissecting aortic aneurysms. Auto-dominant.
Cystic Fibrosis
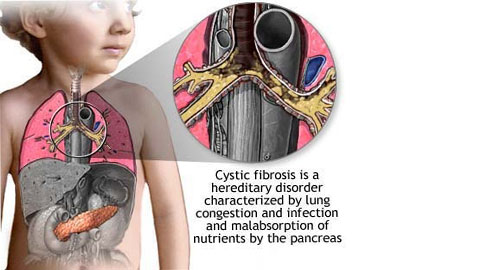
Recessive defect in CFTR gene on chromosome 7. CFTR secretes Cl. Defect causes thick mucus, recurrent infections, nasal polyps, and meconium ileus in newborns. Most common lethal disease in whites. High concentration of Cl found in the sweat test. Treat with N-acetylcysteine to loosen mucus.
Duchennes
X-linked frameshift mutation deletes the dystrophin gene and accelerates the breakdown of muscle. Onset before age 5. Big calfs due to fat replacement. Weak pelvic girdle.
Fragile X syndrome
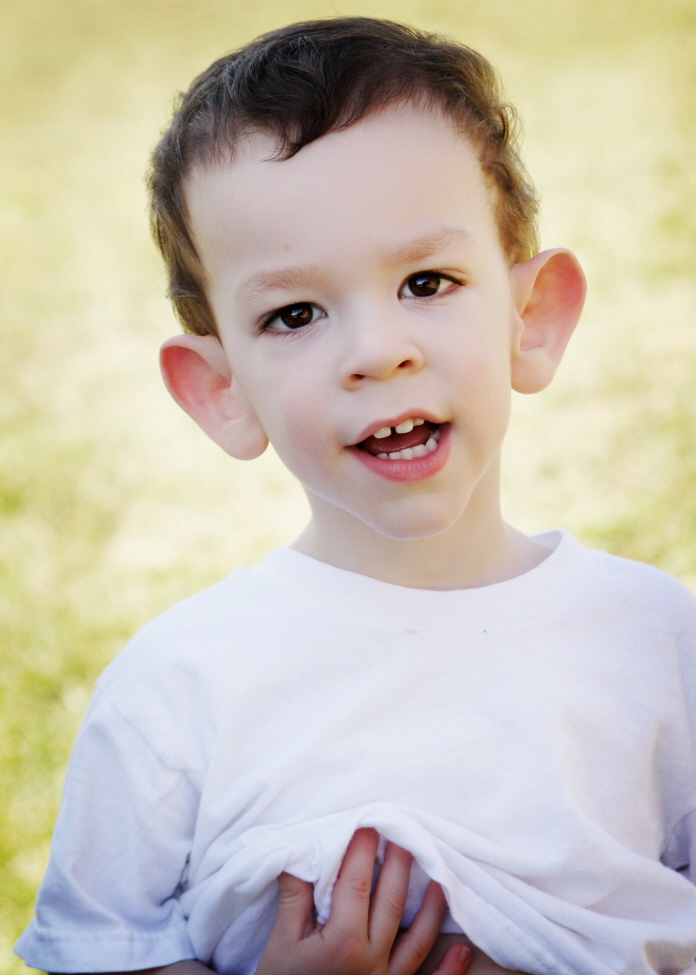
Effects FMR1 gene. Trinucleotide repeat of CGG. Large jaw, large ears, autism. 2nd leading cause of genetic mental retardation.
Down syndrome

Trisomy 21. Most common cause of genetic mental retardation. Meiotic nondisjunction. Associated with congenital heart disease and early onset of Alzheimers.
Edwards syndrome
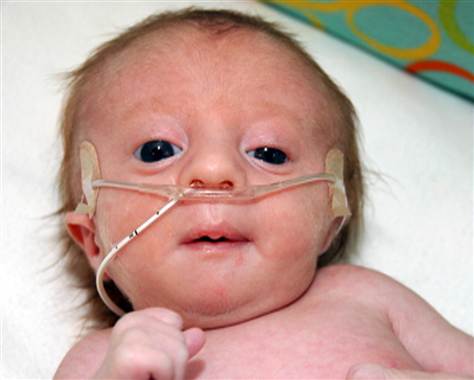
Trisomy 18. 2nd most common live birth trisomy. Death within first year. Clenched hands, rocker-bottom feet, low-set ears, small jaw, congenital heart disease.
Pataus syndrome
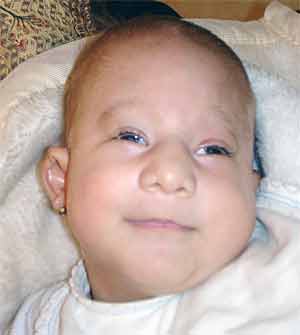
Trisomy 13. Death within first year. Often presents with cleft lip, microphthalmia,congenital heart disease, and holoprosencephaly (forebrain fails to divide into hemispheres - often resulting in cyclops)
Cri-du-chat syndrome
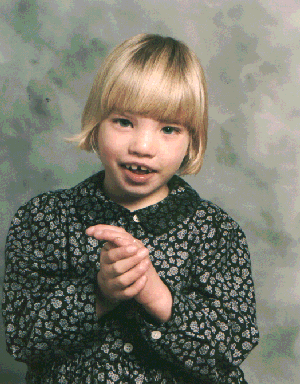
Microdeletion of short arm of chromosome 5. Results in patient giving high pitched meowing cries. Presents as microcephaly, severe mental retardation, epicanthal folds, and cardiac abnormalities.
Williams syndrome

Microdeletion of long arm of chromosome 7. Loss of elastin gene. Distinctive "elfin" face. Good verbal skills and very very friendly. Love music. Cardiovascular problems and some intellectual disability. Hypercalcemia and sensitivity to vitamin D.
http://www.youtube.com/watch?v=PYMoLmFZ9zE
http://www.youtube.com/watch?v=PYMoLmFZ9zE
Ethanol hypoglycemia
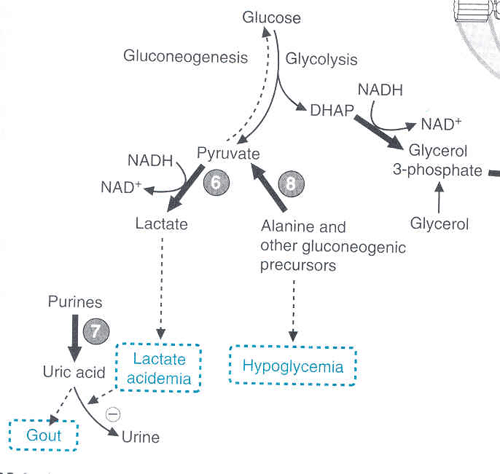
Seen in chronic alcoholics. Results in high NADH resulting in creation of lactate (acidosis) and malate and lots of fatty acid synthesis. There is an inhibition of gluconeogenesis.
Kwashiorkor

Protein malnutrition. Fatty liver develops due to less apolipoprotein synthesis.
Marasmus

Energy malnutrition. Results in wasting and edema.
G-6-P dehydrogenase deficiency
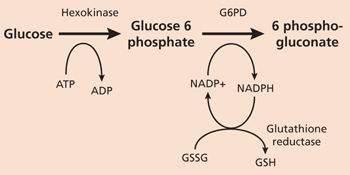
This is the most common enzyme deficiency with high prevalence among blacks. Infers malaria resistance. Results in low NADPH levels. Develop Heinz bodies and Bite cells which are RBCs which have precipitate in them and that have been phagocytosed by spleen macrophages. Fava beans, sulfonamides, primaquine, and anti-TB drugs should all be avoided.
Essential fructosuria
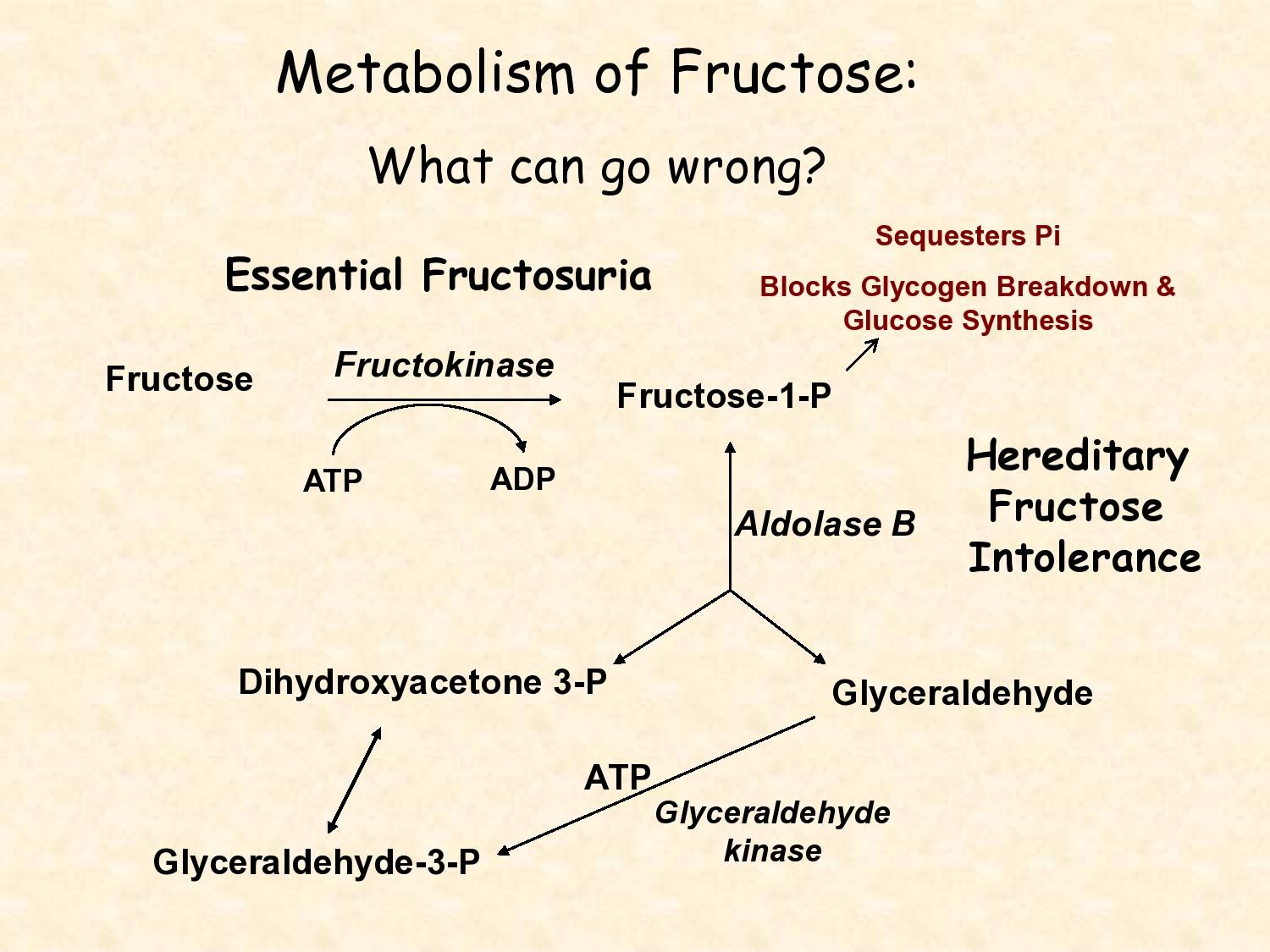
A recessive defect in fructokinase. Frucots appears in blood and urine but the condition is benign and asymptomatic.
Fructose intolerance
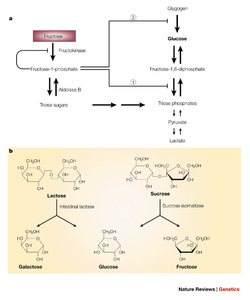
Recessive deficiency of aldolase B. Fructose-1-P accumulates causing a decrease in phosphate which inhibits gluconeogenesis and glycogenolysis.
Symptoms include: jaundice, cirrhosis, vomiting, and hypoglycemia.
Decrease fructose intake.
Symptoms include: jaundice, cirrhosis, vomiting, and hypoglycemia.
Decrease fructose intake.
Classic galactosemia
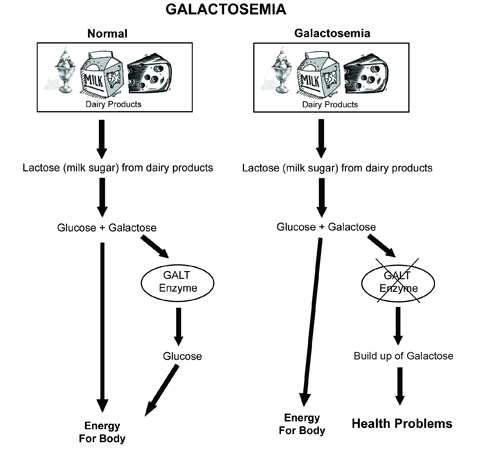
No galactose-1-phosphate uridyltransferase. Auto recessive. Galactitol accumulates in the eye.
Symptoms are failure to thrive, jaundice, hepatomegaly, and infantile cataracts as well as mental retardation.
Tx: exclude galactose and lactose from diet
Symptoms are failure to thrive, jaundice, hepatomegaly, and infantile cataracts as well as mental retardation.
Tx: exclude galactose and lactose from diet
Lactase deficiency
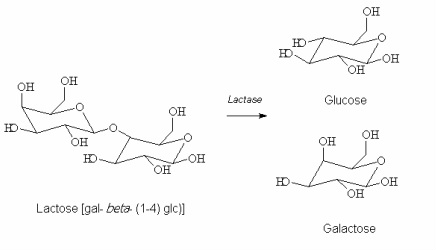
"Lactose Intolerant". Loss of brush-border enzyme. Can be treated with lactase pills.
Hyperammonemia
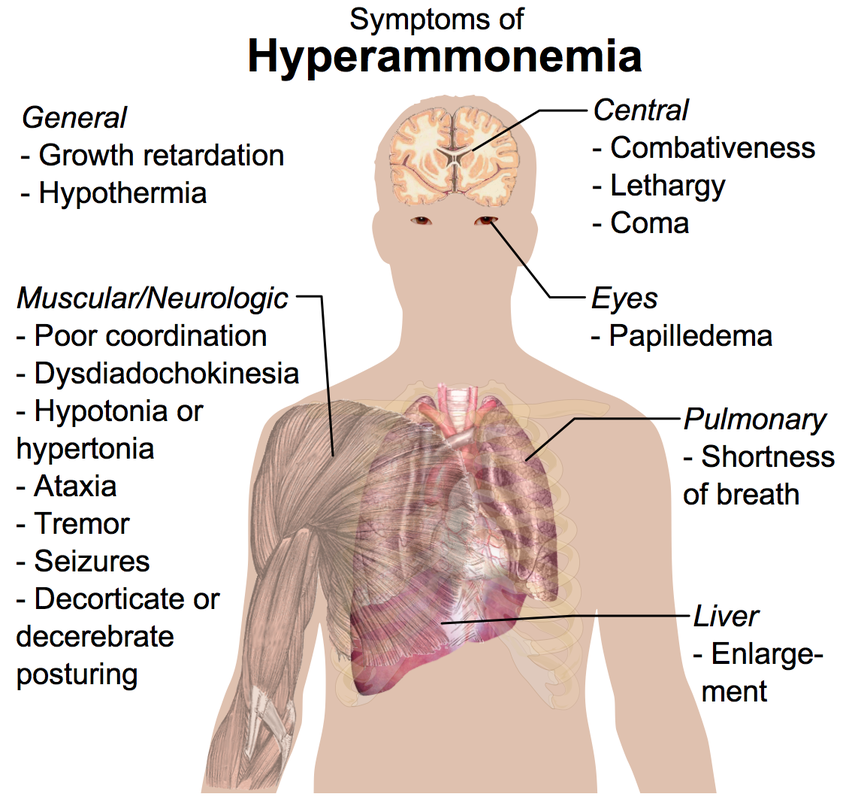
Excess NH4+ which inhibits TCA cycle.
Symptoms: tremors, slurred speech, vomiting, blurred vision
Tx: limit protein, Benzoate or Phenylbutyrate (bind NH4), Lactulose traps NH4 in GI tract.
Symptoms: tremors, slurred speech, vomiting, blurred vision
Tx: limit protein, Benzoate or Phenylbutyrate (bind NH4), Lactulose traps NH4 in GI tract.
Phenylketonuria

Due to low phenylalanine hydroxylase. Tyrosine becomes essential. Auto-recessive or may occur during pregnancy effecting the fetus (microcephaly, heart defects). Leads to a musty body odor, mental retardation, seizures, fair skin, eczema.
Tx: Lower phenylalanine consumption (found in aspartame) and increase tyrosine in diet.
Tx: Lower phenylalanine consumption (found in aspartame) and increase tyrosine in diet.
Albinism
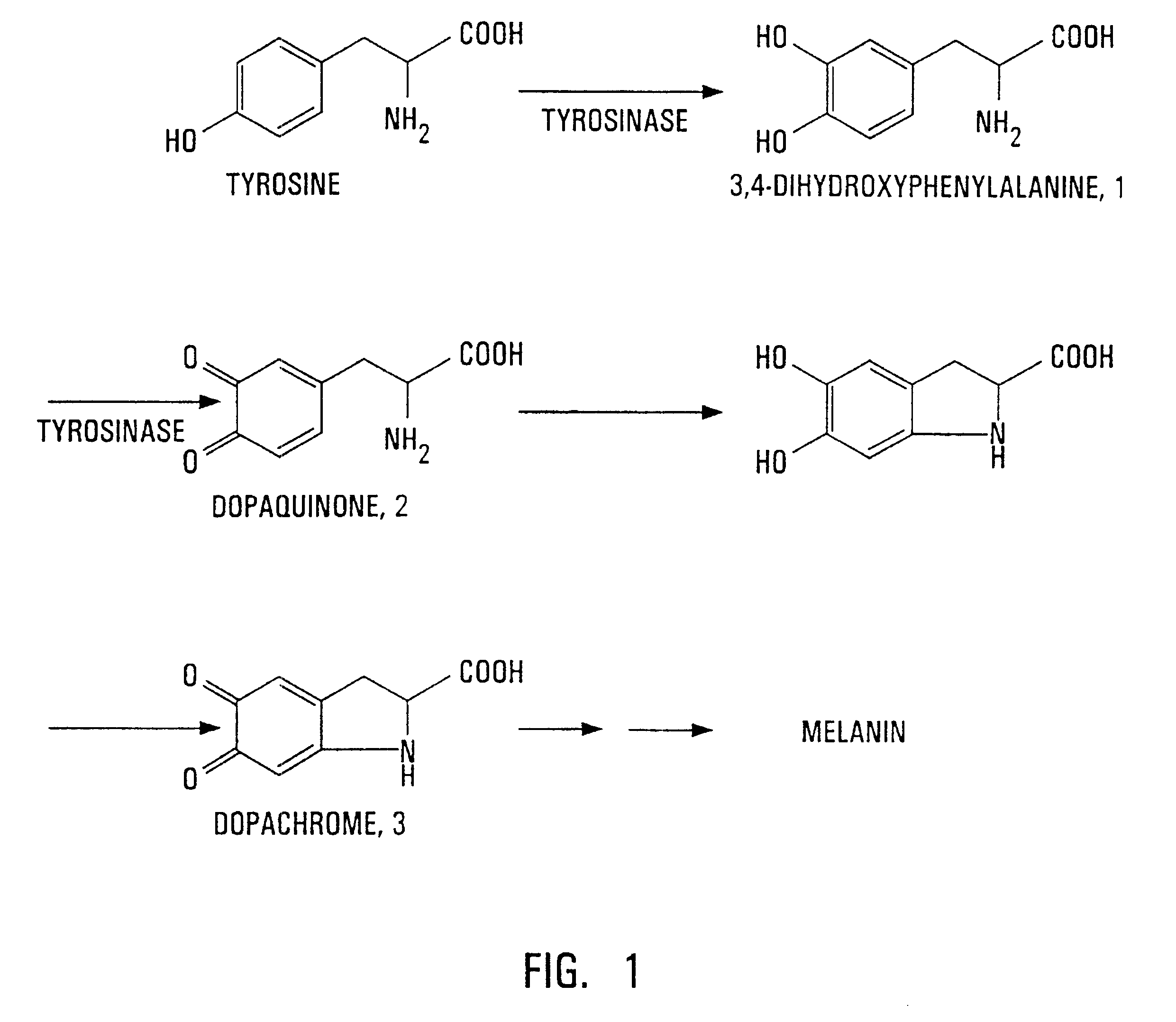
No melanin which leads to skin cancer due to autosomal recessive tyrosinase deficiency or defective tyrosine transporter. Also can happen when neural crest cells fail to migrate.
Homocystinuria
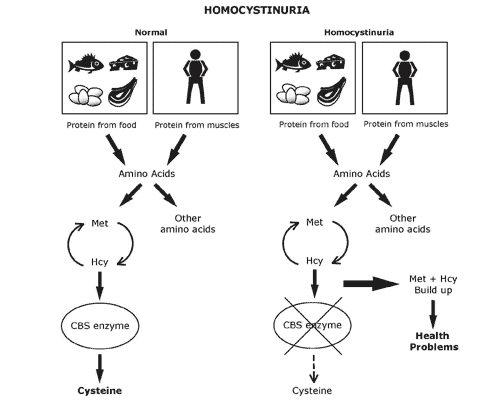
Results in excess homocysteine and cysteine becomes essential. Homocysteine will be found in urine and result in mental retardation, osteoporosis, tall stature, kyphosis. There are 3 auto-recessive forms.
Cystinuria
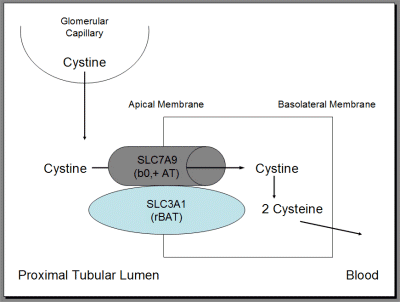
Common auto-recessive defect of renal tubular amino acid transporter of cysteine and others in the proximal convoluted tubule. The excess cysteine in urine precipitates as hexagonal crystals and renal staghorn calculi.
Maple syrup urine disease
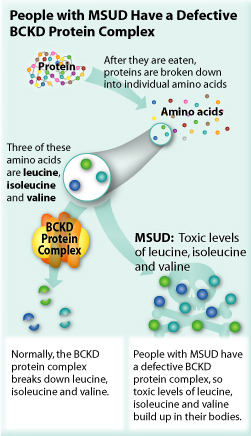
Auto-recessive blocked degradation of branched amino acids Ile, Leu, and Val from low alpha-ketoacid dehydrogenase (B1). Causes severe CNS defects and death.
GLYCOGEN STORAGE DISEASES - abnormal glycogen metabolism resulting in glycogen accumulation.
Von Gierke's disease (type 1)
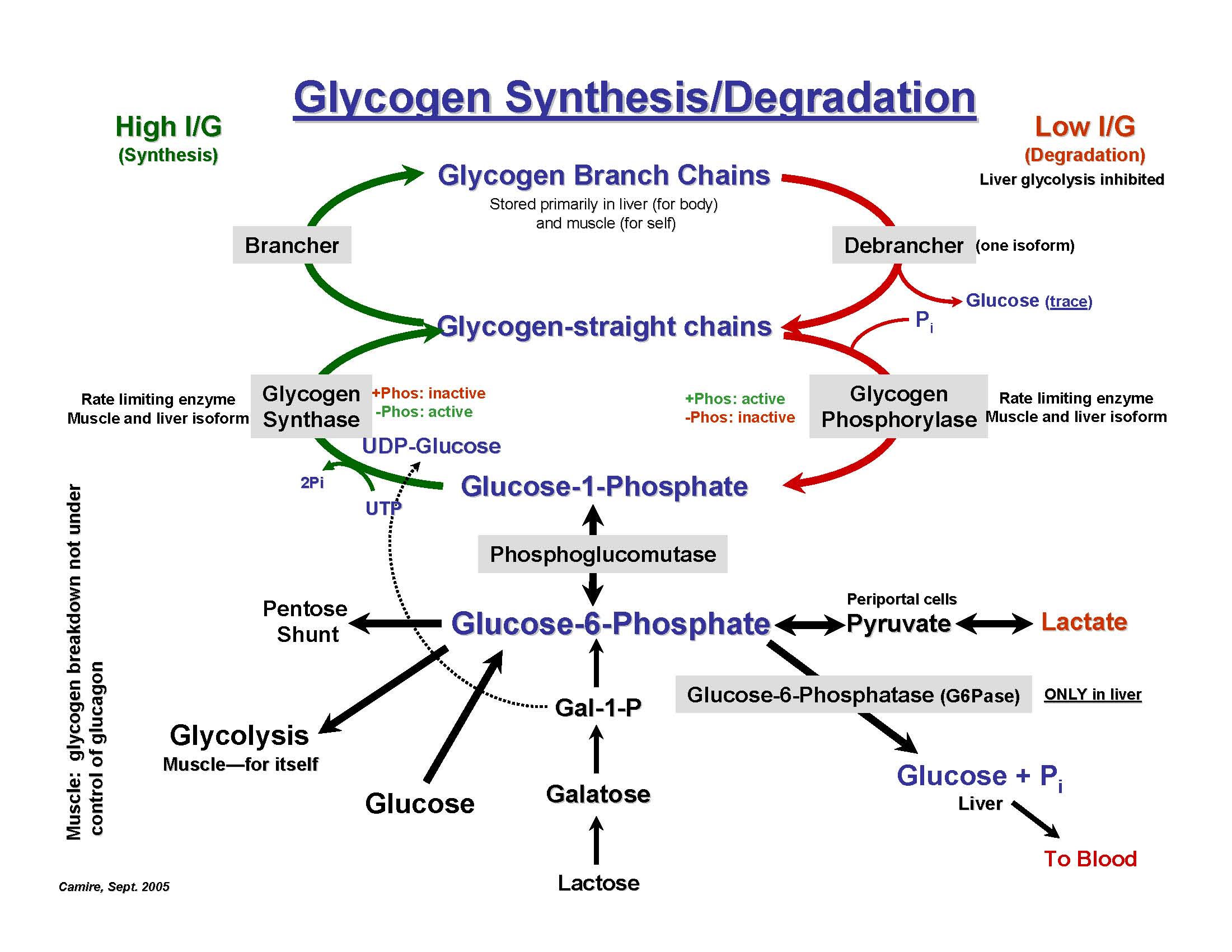
Autosomal recessive deficiency of glucose-6-phosphatase. There is severe fasting hypoglycemia, lots of glycogen built up in the liver and hepatomegaly. Blood lactate levels are low.
Pompe's disease (type 2)

Auto-recessive Lysosomal alpha-1,4-glucosidase deficiency. Patient will have cardiomegaly and early death.
Cori's disease (type 3)
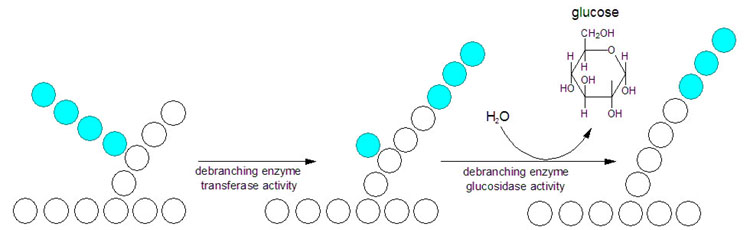
Deficient debranching enzyme. So, similar to type 1 but a less severe. Blood lactate levels are normal.
McArdle's disease (type 5)
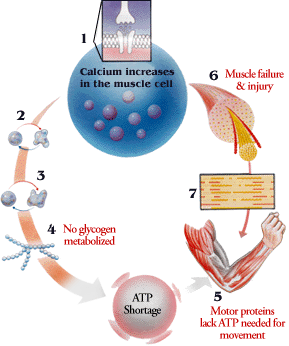
Deficiency in glycogen phosphorylase in muscle. So, there is a lot of glycogen in the muscle which gives you cramps and myoglobinuria with exercise.
LYSOSOMAL STORAGE DISEASE - deficiency in one of many lysosomal enzymes resulting in accumulation of metabolic product.
Fabry's disease
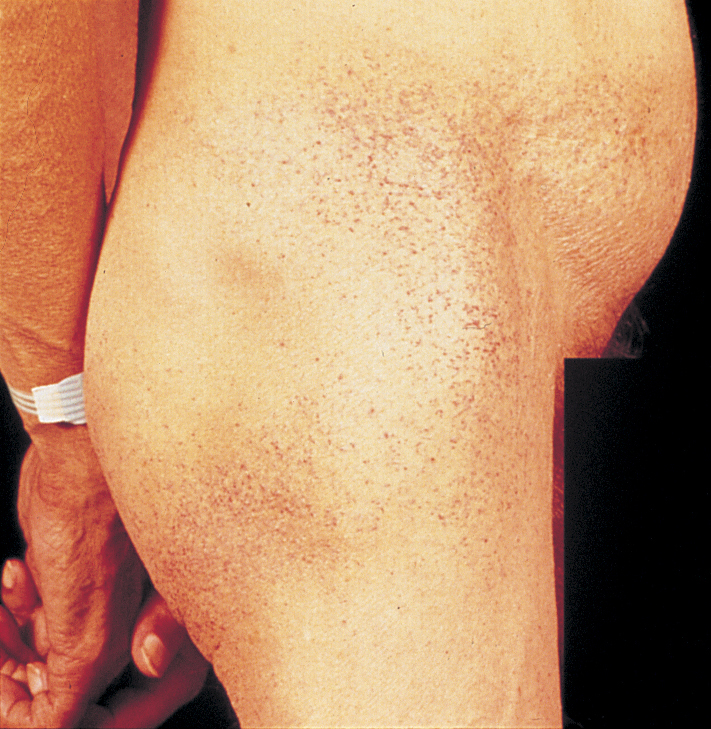
X-linked recessive alpha-glactosidase deficiency resulting in ceramide trihexoside accumulation. Patient has peripheral neuropathy of hands and feet, angiokeratomas, and cardiovascular and renal disease.
Gaucher's disease

Auto-recessive glucocerebrosidase deficiency with accumulated glucocerebroside. It is the most common lysosomal storage disease. Gaucher cells are macrophages that look like crumpled tissue paper. Patient will also have hepatosplenomegaly and aseptic necrosis of femur.
Niemann-Pick disease
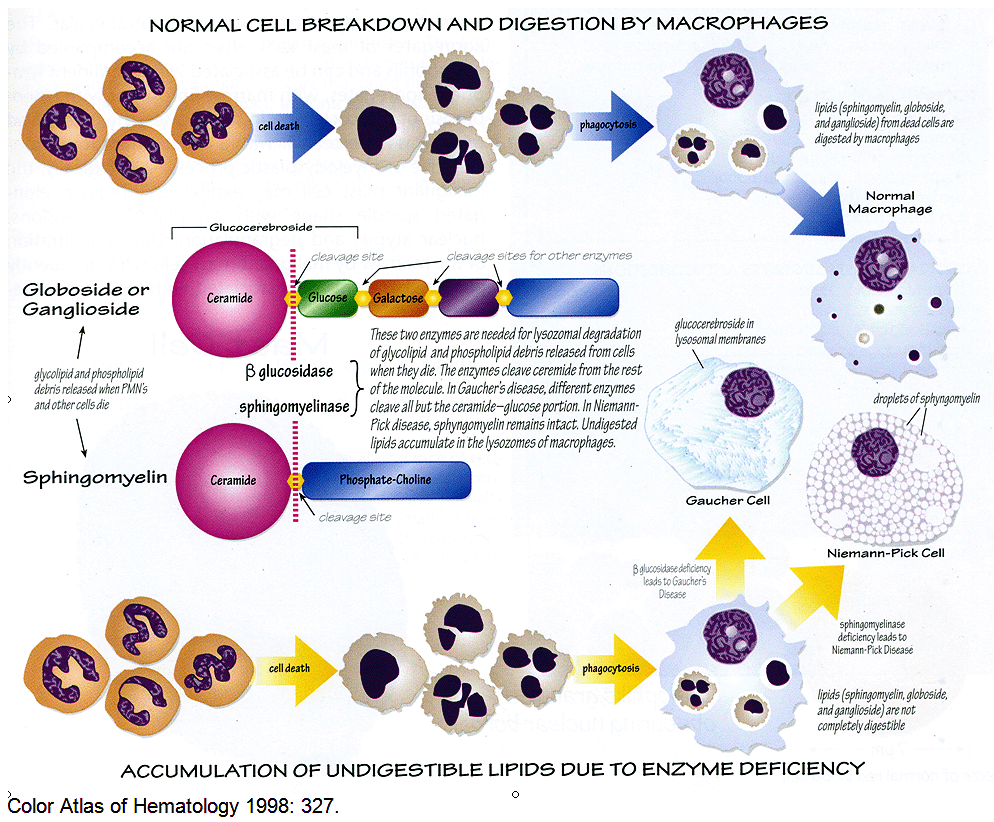
Auto-rec Sphingomyelinase deficiency with accumulating Sphingomyelin causing neurodegeneration, cherry-red spot on the macula, and foam cells.
Tay-Sachs disease

Auto-recessive deficiency of hexosaminidase A resulting in lots of GM2 ganglioside. Manifests as neurodegeneration, cherry-red spot on macula, lysosomes with onion skin appearance, and no hepatosplenomegaly.
Krabbe's disease

AR Galactocerebrosidase with accumulation of Galactocerebroside manifesting as peripheral neuropathy, developmental delay, optic atrophy and globoid cells.
Judson: http://www.youtube.com/watch?v=DzuvW3pgZLM
Judson: http://www.youtube.com/watch?v=DzuvW3pgZLM
Metachromatic leukodystrophy
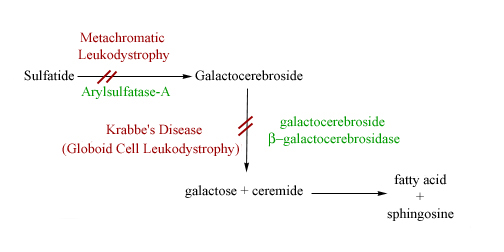
AR deficiency of Arylsulfatase A resulting in accumulation of Cerbroside sulfate. Presents with central and peripheral demyelination with ataxia and dementia.
Sam: http://www.youtube.com/user/keysgetaway/videos
Sam: http://www.youtube.com/user/keysgetaway/videos
Hurler's syndrome

AR deficiency of alpha-L-iduronidase leading to accumulation of heparan sulfate and dermatan sulfate causing developmental delay, gargoylism, airway obstruction, corneal clouding, & hepatosplenomegaly.
Hunter's syndrome

XR deficient Iduronate sulfatase with accumulation of Heparan sulfate and dermatan sulfate. It is a milder form of Hurler's with the addition of aggressive behavior and absent corneal clouding.
http://www.youtube.com/watch?v=odUrHuIRD2s
http://www.youtube.com/watch?v=odUrHuIRD2s
Familial dyslipidemias
Type I hyperchylomicronemia
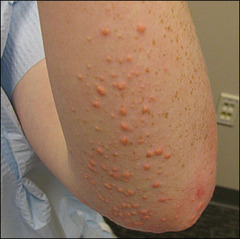
AR deficiency in lipoptoein lipase or altered apolipoprotein resulting in increased levels of chylomicrons, TG, and cholesterol. Manifests with pancreatitis, hepatosplenomegaly, and eruptive xanthomas without a significant risk for atherosclerosis.
Type IIa familial hypercholesterolemia
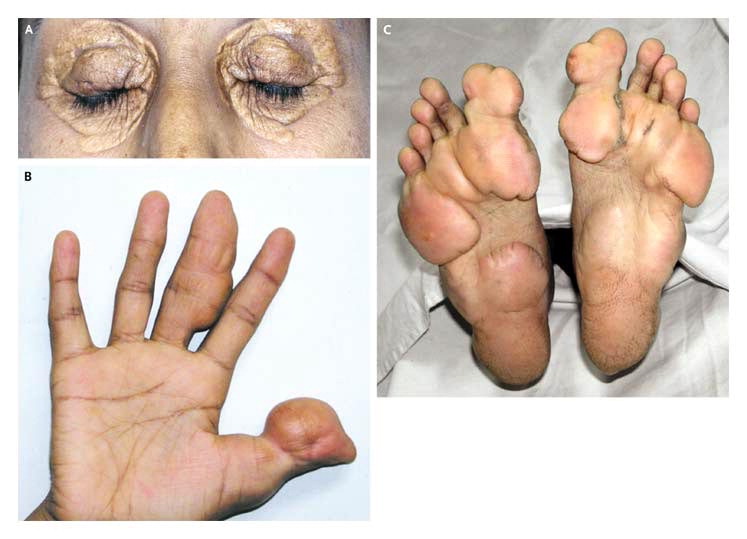
AD absent or low levels of LDL receptors causing atherosclerosis, tendon xanthomas, and corneal arcus. Lots of LDL and cholesterol seen in blood.
Type IV hypertriglyceridemia
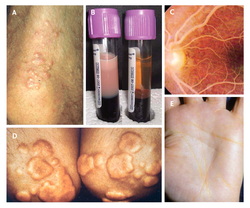
AD hepatic overproduction of VLDL causing pancreatitis. Increased levels of VLDL and TG found in blood.
